Normalizacja danych
Biorąc pod uwagę iż macierze zostały stworzone w celu analizowania między innymi genotypów, wymusza to wykonywanie operacji na bardzo dużej ilości danych, generowanym w pojedynczym eksperymencie, dlatego wymusza to zastosowanie odpowiednich narzędzi statystycznych.
Plan normalizacji danych:
- obliczenie ilorazu intensywności fluorescencji :
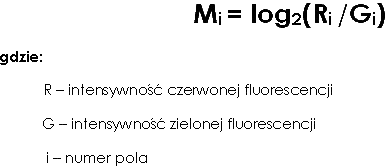
Zastosowanie logarytmu o podstawie 2 powoduje, że dwukrotny przyrost poziomu ekspresji odpowiada Mi = 1 a dwukrotny spadek poziomu ekspresji spada do Mi = -1
- Obliczenie średniej intensywności fluorescencji pola w skali logarytmiczej:
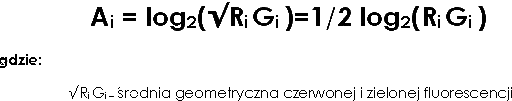
Dzięki wynikom uzyskanym z obliczenia tego wzoru, otrzymamy z zebrania tych punktów wykres ukazujący jak układa się zróżnicowanie ilorazu w naszych próbkach. Skupiska mówią nam o geny o takiej samej albo bliskiej ekspresji (bierzemy pod uwagę błędy wynikające min. Przygotowania próbki, niechcianego tła itp.). Osoby analizujące dane powinny skupić się na izolowanych punktach, gdyż one reprezentują geny, w których ekspresja uległa zmianie(zwiększeniu/zmniejszeniu). Jednakże przed pełną analizą wyników należy postarać się o usunięcie jak największej ilości błędów, które mogły zaistnieć podczas eksperymentu – i tak tez przechodzimy do normalizacji naszych pomiarów.
Zauważmy jednak wcześniej kilka prawidłowości wynikających z danych, a które są bardzo dla nas ważne, a mianowicie tworząc próbki testowe oraz próbkę referencyjną staramy się aby całkowita ilość cDNA z każdej z próbek była taka sama. Jeżeli ekspresja genu nie uległa zmianie to oczywistym jest fakt iż Ri =Gi oraz M=0.
(techniki normalizacji globalnych)
Jeżeli jednak nasz wykres umiejscowił się znacznie poniżej linii 0, należy się spodziewać, że dane zostały obarczone błędem systematycznym. Jednym ze sposobów eliminacji tego błędu jest normalizacja danych względem genów o ekspresji konstytutywnej(ang. Housekeeping genes), czyli genów dla których zakłada się stały poziom ekspresji(nie wynikły żadne różnice pomiędzy próbka testową a próbką referencyjną). Wykorzystując tą wiedzę, staramy się umiejscowić wartości odpowiadające tym genom jak najbliżej 0 , czyli aby nasze Mi =0. Jednakże największym problemem w tej metodzie jest ustalenie, których genów ekspresja faktycznie nie uległa zmianie. Również dochodzą problemy związane z tłem itp. Dlatego alternatywnym sposobem, znacznie łatwiejszym jest dodanie pewnej stałej do wartości Mi , której zadaniem będzie przesunięcie wykresu tak aby jego średnia znajdowała się w punkcie zero. Te dwie metody normalizacji są technikami globalnymi, działającymi na wszystkich punktach wykresu. Można te metody zastosować również zakładając, że nie występuje błąd wynikający ze znacznego zróżnicowania wydajności znakowania poszczególnych próbek. Jednakże jeżeli chcemy dokładniej zbadać i zminimalizować możliwy błąd możemy przeprowadzić dodatkowy eksperyment, który polega na zamianie czerwonego barwnika z zielonym i obliczenie ich średniej. Wartości Mi obu eksperymentów odejmuje się od siebie uzyskując dwukrotność prawidłowej wartości Mi . Metoda ta jest nazywana metodą samonormalizacji (ang. Self-normalization). Pozwala ona wyeliminować każdy typ błędu, gdzie może wyniknąć niezamierzona zmienność pomiędzy poszczególnymi polami. Jednakże plusem a zarazem minusem jest fakt iż działa ona globalnie optymalizując wszystkie dane. Ale w rzeczywistości nie koniecznie wszystkie trzeba normalizować.
(techniki normalizacji lokalnych)
Niejednokrotnie występują błędy związane z odczytem danych przez detektor fluorescencji, którego czułość została w jakiś sposób zaburzona. Błędem który również będzie podawał podobne rezultaty jest błąd nieliniowości procesu hybrydyzacji znakowanego DNA do pól. Aby wyeliminować tego typu błąd można zastosować technikę normalizacji lokalnie wyważonego wygładzenia wykresu rozrzutu. (ang. Locally Weighted Scatterplot Smoothing, LOWESS). Polega ona na dopasowaniu do punktów pomiarowych funkcji m(F), która będzie reprezentować gładką krzywa. Następnie wyznacza się skorygowane wartości ilorazów intensywności : Mi - m(Fi). Zauważmy, że wartości m(F) zależą od wszystkich punktów pomiarowych pochodzących z pól o intensywności F, co powoduje że ta technika jest odmienną od wcześniej przedstawionych.
Skoro już wiemy jak czytać dane, oraz poddawać je normalizacji w celu usunięcia przypuszczalnych błędów. Należy zadać sobie pytanie jak się upewnić, które zmiany poziomów ekspresji nie są dziełem przypadku. Przydatnym tutaj wyjściem będzie zastosowanie wzoru na wyliczenie wartości średniej ? oraz odchylenia standardowego ? z ilorazów intensywności M. Mając te wartości należy je zastosować do następującego wzoru: Z=(M - ?)/? Gdy rozkład wartości M jest zgodny z rozkładem normalnym, to prawdopodobieństwo wystąpienia wartości Z spoza przedziału -1.96